
There are parts of human DNA that are of viral origin: many of them were inserted into the primordial genetic material of our ancestors many millions of years ago and have been inherited by successive generations ever since. Human endogenous retroviruses (HERV) are by far the most common virus-derived sequences in our genome. Most HERV sequences have long been assimilated and therefore are shared by all individuals in the human population, but not all are and a few have been known to be found in only a subset of individuals. Most of these unfixed HERV elements are known to descend from relatively recent insertion events that are still segregating in the human population. But new research recently published in Mobile DNA shows that another mechanism has introduced more inter-individual variation in HERV content between humans than previously appreciated. How can this be?
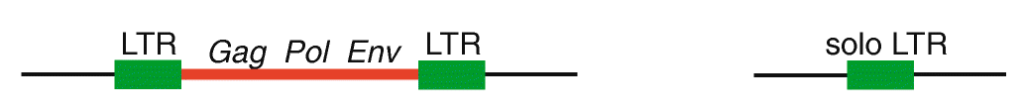
First, it’s important to think about the structural features of HERVs. To be integrated in the host chromosome, these sequences must be full-length elements called proviruses. Each provirus is organized around a central core containing the viral coding genes sandwiched between long noncoding sequences repeated on each end called long terminal repeats (LTRs) (see Figure 1). Following integration, the two LTRs of a provirus, which are identical at the time of insertion, frequently recombine to form what is referred to as a solo LTR. The recombination process eliminates internal viral genes along with one of the two LTRs, leaving behind a single LTR. It has been estimated previously that 90% of all HERVs in the human genome are solo LTRs, and only 10% remain in their proviral form. But what if some of these proviral elements are still undergoing the transition to becoming solo LTRs? Researchers at the University of Utah and at Cornell University set up to investigate this question and assess the extent by which the process of LTR recombination could generate HERV variation among humans.
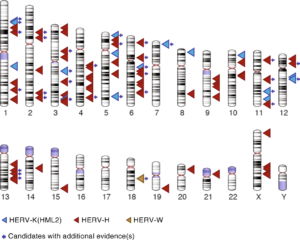
Dr. Jainy Thomas developed a new computational approach that would allow them to screen large amount of DNA sequences from diverse human populations to find what presumably would be rare LTR recombination events. Given the vast amount of HERV sequences in human genomes, the task was akin to finding needles in a haystack. A publicly available data set, supported by the Simons Foundation, of whole genome sequences representing 130 different genetic populations were searched for variants from three different retroviral families: HERV-K(HML2), HERV-W and HERV-H. The pipeline she developed allowed Dr. Thomas to recover most of the HERV variants previously cataloged and discover many more (Figure 2). Perhaps not surprisingly, most of the newly discovered variants were apparently rare, as they were found in just one or a few individuals. But they were also unexpected given that many of these HERVs had been inserted long ago in the DNA of our ancestors and some were even shared with our great ape relatives, and thus thought to be fixed in the human population. Nonetheless, Dr. Thomas was able to confirm experimentally that several of these variants do segregate in the human population, thereby validating the efficiency of her computational approach.
Does this type of HERV variation matter? There are many reasons to think that these genetic variants could represent an overlooked source of physiological variation between humans that contribute to disease susceptibility. Indeed, there is mounting evidence that the three HERV families investigated in the study exert both beneficial and pathogenic effects on humans. For instance, HERV-encoded genes are overexpressed in patients affected with diseases, including Amyotropic Lateral Sclerosis (ALS, or Lou Gehrig’s Disease), Multiple Sclerosis and several cancers, and their gene products are thought to be implicated in the etiology or progression of these diseases. On the other hand, some HERVs appear to have beneficial properties. Recently, Dr. Feschotte and his colleagues showed that some HERVs are critical for the proper regulation of the human immune response. Proviral HERV-H elements, one of the sources of variants revealed in the new Mobile DNA study, have been shown to be important for embryonic cells to retain their pluripotency (i.e., their ability to differentiate into diverse cell types). Thus, better quantifying the presence or absence of these proviral HERVs across individuals and populations will be important to better understand how these elements affect human health and physiology.

Rebecca Pearce
Latest posts by Rebecca Pearce (see all)
- Placental Sex Differences: Q&A with Amy Inkster - 9th September 2019
- Transposon Day Quiz: 2019 - 10th June 2019
- World Penguin Day quiz - 24th April 2019
Comments