How much of a problem is still posed by MRSA in healthcare settings?
The history of methicillin-resistant Staphylococcus aureus (MRSA) is one punctuated by waves of successful clones that spread and are subsequently replaced by others. MRSA pose a continuing threat due to the ebb and flow of the population.
Although the situation in the UK has greatly improved since 2004, MRSA remains a major scourge in healthcare settings globally. In Singapore, the organism is the most common antimicrobial-resistant bacterium causing nosocomial infections, with more than 3,000 infections logged each year in a population of 5 million.
As our study has demonstrated, the burden of disease is caused by successful clones that have adapted to surviving and thriving in hospital settings. This means they are particularly tenacious and difficult to eradicate, therefore posing a constant threat to vulnerable patients.
In addition to the problem that MRSA poses in healthcare settings, epidemic clones of MRSA emerged in recent years that cause disease in the community, and can also spread into hospitals. This demonstrates the blurred lines between what we consider to be healthcare- and community-infections, and also the adaptability of MRSA as a human pathogen.
Why did you focus on Singapore in this particular study?
This study reflects the combined interests of the groups in the UK and Singapore of gaining a greater understanding of this success of MRSA, and improving the ways in which to combat it.
The seed of this collaboration was sown by a sabbatical that brought Dr Li Yang Hsu, an infectious disease physician from Singapore, to the Wellcome Trust Sanger Institute in Cambridge, to work alongside Dr Matt Holden in the Pathogen Genomics group, and gain experience in bioinformatics and whole genome sequencing.
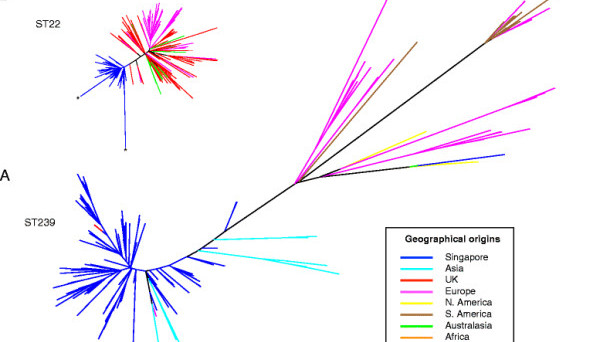
…the succession of MRSA in Singapore provided a unique opportunity to look at two clones going head-to-head in a healthcare system.
Previously the application of whole genome sequencing to study the emergence and evolution of bacterial pathogens has tended to focus on individual clones, and so the succession of MRSA in Singapore provided a unique opportunity to look at two clones going head-to-head in a healthcare system.
In many respects Singapore proved be an ideal study location for investigating the evolutionary success of a MRSA; Singapore is a small country with a closely interlinked public hospital system, where an older dominant MRSA clone had rapidly been losing its ‘monopoly’ to a newer pandemic MRSA clone that had originated in the UK.
How difficult was it to obtain samples from a 30 year period for this work?
Because clinical laboratory space is at a premium in densely populated Singapore, it is the practice of local laboratories to discard all their clinical bacterial isolates after a prescribed period of time, unless such isolates were kept for ad-hoc research or other projects. In this study we were fortunate that the local hospitals had kept isolates from the 2000s, mainly due to the efforts of local clinicians whose interest in the growing problem of MRSA had ensured that the isolates were retained.
The older isolates proved to be a bigger problem, but here we were again extremely lucky that an older collaboration with Prof Warren Grubb from the Gram-Positive Typing Laboratory at Curtin University, Perth, Australia had resulted in Singaporean isolates from the 1980s and 1990s being stored over there.
You utilised Bayesian techniques for the study of the history of MRSA in Singapore, what advantages do these have?
In this study we used a series of methods and tools to investigate the genetics and evolutionary relationships of MRSA in Singapore, including some in which Bayesian statistical methods lay at their heart.
Using these techniques we were able to delve into the whole genome sequencing data to gain a greater temporal understanding of the evolutionary events that have shaped the MRSA populations, thereby highlighting significant events in the history of MRSA in Singapore. We were also able to use these methods to examine the genetic diversity of competing MRSA clones over time, and pinpoint notable changes in the populations.
Ultimately it was the resolution that the Bayesian techniques provided for analyzing the whole genome sequencing data that proved to be most advantageous. Not only did it help identify the emergence of a new MRSA variant, but it also identified trends in the data that were supported by the subsequent epidemiological observations.
One of your main findings is that antibiotic usage has had an important role in driving the evolution of methicillin resistance. Is there evidence that Staphylococcus aureus is also becoming resistant to alternative antibiotics?
On the contrary, the newer epidemic clones of MRSA are becoming less resistant to alternative, non-b-lactam antibiotics; methicillin being a b-lactam antibiotic. It is plausible that being resistant to multiple antibiotics comes with a fitness cost, and the newer clones of MRSA have evolved to be more efficient in this regard.
In our study, and earlier published works looking at several global MRSA clones, the key antibiotic resistance determinant (other than to b-lactam antibiotics) is to the fluoroquinolone antibiotics, a class of antibiotics that are excreted in sweat and on to the skin. The increase in the use of fluoroquinolones in recent decades has been a key driver of the success of pandemic clones of MRSA that have developed resistance to this antibiotic.
You report that early Singaporean MRSA strains correspond to those seen in the Hungarian epidemic, and that MRSA strains that appeared in Singapore from the year 2000 onwards corresponded to those seen in UK hospitals in the early 1980s. Does this turn the common fear of diseases spreading into the West on its head?
This merely shows how interconnected the world has become. Incidentally, in this study, we also discovered that one of the MRSA strains that had caused a big outbreak in a London hospital in the 2000s had likely originated from Singapore.
A recurring observation from many of the whole genome sequencing studies examining the emergence and spread of bacterial human pathogens is that the pathogen’s population structure is as much a story about humans and their activities, as it is about bacteria.
Multiple pandemic clones of MRSA have emerged over the years that circulate globally.
For MRSA, an organism that colonizes the skin and has adapted to survive and thrive in hospitals, this is particularly evident. Multiple pandemic clones of MRSA have emerged over the years that circulate globally, driven by a dynamic population (and also their antibiotic usage). In this regard Singapore occupies an exquisite position as far as the epidemiology of infectious disease is concerned, being a densely-populated global hub.
What does your study mean for research into MRSA and other pathogen epidemics?
This study has highlighted that, alongside clinical practice and antibiotic usage, competition between bacterial populations in a healthcare setting has an important role in driving pathogen evolution.
In particular, it revealed that during clone succession, new variants emerged in a declining clone via horizontal gene transfer, introducing genes that promote survival and persistence. In the case of the MRSA we have studied, the genes in question comprised an arginine catabolic mobile element (ACME), a mobile genetic element that is also found in an epidemic community-associated MRSA (USA300) that has become dominant in the USA.
The functions encoded by ACME are linked to enhanced skin colonization, and highlight than an important facet of the success of MRSA is its ability to interact with the largest organ on the human body.
More generally, our study illustrates the value of examining the success of epidemic clones in a wider context rather than in isolation. This is especially important from a public health point of view. As sequencing technology becomes more widely embraced into pathogen surveillance, we will have the capacity to take a more holistic approach to investigating pathogen population structure. With the right tools and approaches we will be able to use whole genome sequencing to not only monitor present dangers, but to identify where new problems emerge out of previous dangers, and target our interventions accordingly.
What is next for your research?
The next chapter for this particular work is a collaborative project between Singaporean hospitals, the Wellcome Trust Sanger Institute, and the University of St. Andrews, to examine the transmission of MRSA between acute-care and community hospitals, and also between nursing homes and the hospitals.
More widely, the goal of our future research is to translate the genomic perspective into a physiological understanding of what makes a successful hospital-adapted microorganism.

Sophie Marchant
Latest posts by Sophie Marchant (see all)
- Happy Easter! Enjoy the sound (and taste) of your chocolate! - 23rd March 2016
- Quiz: Aiming to make rare diseases common knowledge - 29th February 2016
- Quiz: The big end of year On Biology bumper quiz - 23rd December 2015
Comments